Especialistas alertam para a necessidade de conscientização e conhecimento das mucopolissacaridoses (MPSs) e reforçam a importância do diagnóstico precoce por meio da expansão do teste do pezinho e do tratamento mais adequado. O Dia Internacional de Conscientização sobre as MPSs foi comemorado neste mês.
Também chamadas de Síndrome de Hunter ou MPS-II, as MPSs são doenças genéticas raras, sendo o tipo II o mais comum no Brasil. Sua presença é resultado de uma falha em um gene localizado no cromossomo X. Por esse motivo, a síndrome ocorre quase exclusivamente em meninos.
De acordo com o geneticista do Hospital de Clínicas de Porto Alegre Roberto Giugliani, a MPSs é uma doença de depósito. “Nosso organismo é uma usina de reciclagem, sempre aproveitando seus componentes. O mucopolissacarídeo é fabricado no organismo e é importante para a movimentação das articulações e para dar sustentação aos órgãos. É um componente muito presente em várias partes do organismo”, explicou Giygliani, que é também professor da Universidade Federal do Rio Grande do Sul e co-fundador da Casa dos Raros.
Segundo o médico, ao mesmo tempo que o organismo produz essa substância, precisa destruir parte do que foi fabricado. Porém, nas pessoas que têm a Síndrome de Hunter, o corpo não entende que é preciso degradar a substância, gerando então o excesso, que se acumula em diferentes órgãos e causa diversos problemas e desequilíbrio no organismo. “O que causa essa falha no mecanismo de degradação é uma falha na sequência do DNA, alterando a produção da proteína que degrada o polissacarídeo responsável por manter esse equilíbrio. É uma doença não rara, e sim, ultrarrara, porque acomete menos de 1 a 70 mil indivíduos. No nosso laboratório, conhecemos mais de 400 casos, mas é possível que existam mais no Brasil.”
Os sintomas são perceptíveis nos primeiros meses de vida: a criança com MPS-II pode ter aumento do fígado e do baço, articulações enrijecidas, atraso na fala, dificuldade de atenção e perda de habilidades adquiridas, entre outras manifestações. Contudo, esses sinais podem ser confundidos com outras patologias, fazendo com que o paciente passe por diferentes especialistas e seja submetido a uma série de exames, por vezes a tratamentos inadequados, até receber o diagnóstico correto, por meio de testes bioquímicos e genéticos.
“Dependendo do gene que está alterado, temos uma proteína que está alterada, e isso resulta em um quadro mais grave ou mais atenuado. Até dentro no mesmo tipo, existem algumas variações, mas, nos casos mais graves, o paciente que, em geral, nasce com aspecto normal, vai apresentando essa acúmulo progressivo dos mucopolissacarídeos ao longo da vida e quando chega à idade entre 10 e 20 anos com complicações bem grandes. Às vezes, os casos mais graves não sobrevivem à segunda década de vida”, disse Giugliani.
Portadores da Síndrome de Hunter podem apresentar dificuldade para caminhar, devido aos problemas nas articulações e dos ossos, problemas no coração, por causa das válvulas cardíacas, problemas de visão, porque a córnea fica meio opaca, deficiência de audição, porque os ossos do ouvido ficam afetados, e problemas de respiração, porque há dificuldade de colocar o ar para dentro do pulmão.
“A parte neurológica também pode ser afetada, com o indivíduo começando bem a vida e, depois do segundo ou terceiro ano de vida, regredindo neurologicamente, perdendo algumas habilidades que tinha. A parte cognitiva começa a degradar. A maioria dos pacientes tem esse comprometimento neurológico que é, na verdade a parcela que temos a maior dificuldade para tratar com os meios disponíveis atualmente”, acrescentou o médico.
De acordo com Giugliani, não é difícil diagnosticar a doença, embora a maioria dos laboratórios não disponha de exames para o caso. Como é uma doença rara, apenas alguns laboratórios de referência permitem fazer o diagnóstico, “O mais difícil é um médico pensar nessa possibilidade e, uma vez que ele pense na possibilidade, tem que buscar um laboratório que faça esse exame. No entanto, a execução do exame no laboratório não é uma coisa complicada. São feitos dois exames bioquímicos nos quais se busca a substância que está acumulada. Podem ser fazer também exames genéticos.”
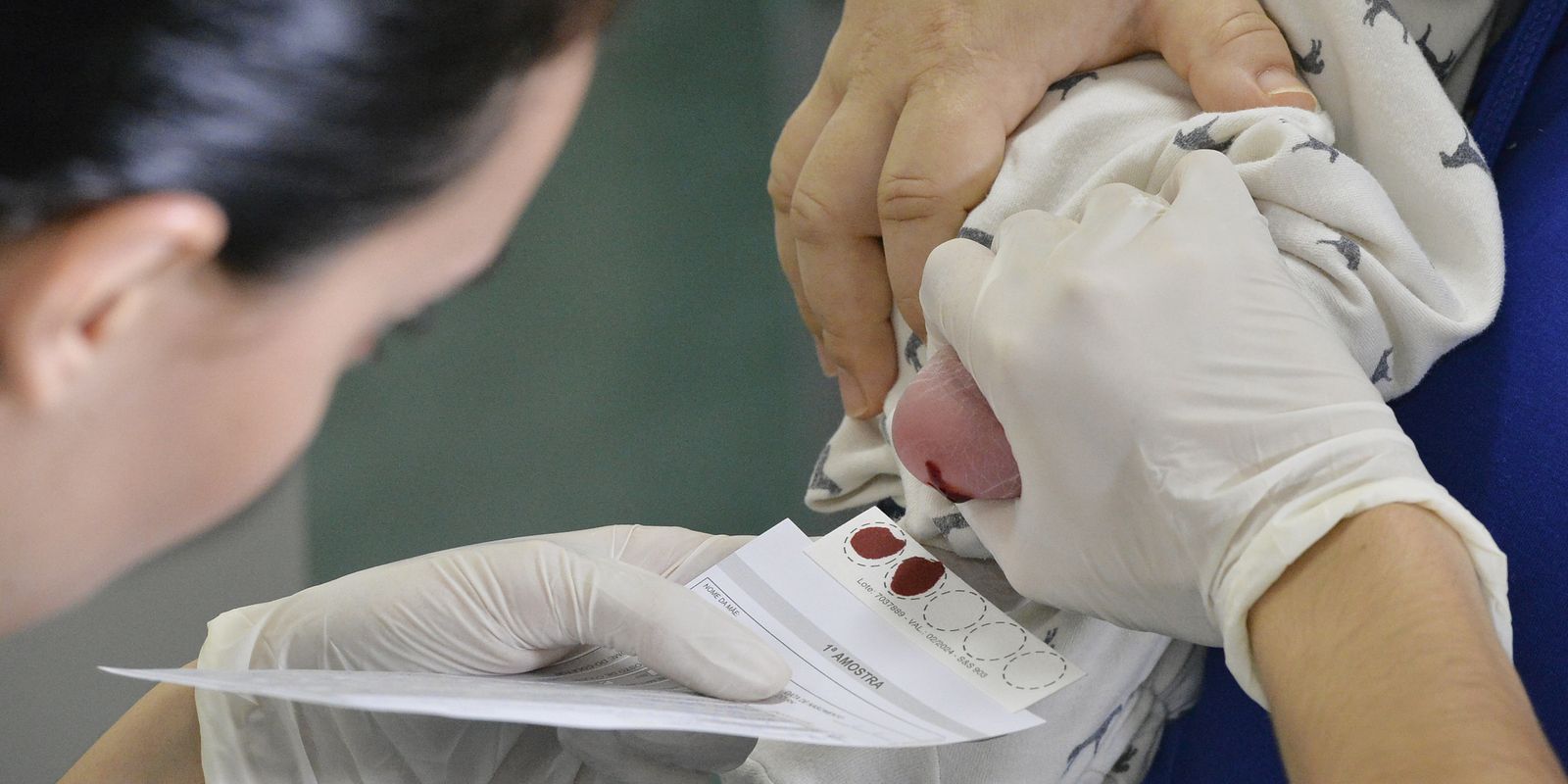
Outra forma de detectar precocemente a a síndrome é o teste do pezinho. A expansão do teste para incluir as MPSs seria uma maneira de detectar rapidamente o tipo II, mais comum no Brasil, além de outras doenças raras, garantindo o tratamento adequado para minimizar os efeitos de tais patologias nos pacientes. O exame já é previsto em lei desde 2021, mas o Brasil ainda enfrenta dificuldades para efetivar sua implantação em toda a rede do Sistema Único de Saúde (SUS).
Novo tratamento
Atualmente, o tratamento disponível no Brasil para a MPS-II não é capaz de atenuar os efeitos neurológicos da doença por causa da chamada “barreira sangue-cérebro”, formada por um conjunto de células que atuam como um filtro altamente seletivo, que protege o sistema nervoso central de ataques de microrganismos e impede que medicamentos administrados por via oral ou injetados no sangue cheguem até o cérebro.
“Aos 3 anos de idade, meu filho foi diagnosticado com mucopolissacaridose II. O medicamento disponível no Brasil não conseguia controlar a piora no quadro neurológico. Depois de pesquisas incessantes conheci esse tratamento inovador e, desde então, venho atuando no convencimento junto à Anvisa [Agência Nacional de Vigilância Sanitária] para que o tratamento seja uma realidade para todos os pacientes com MPS II, e não apenas o meu filho, que está participando da pesquisa clínica”, contou Antoine Daher, pai de Anthony e presidente da Casa Hunter, fundada por ele para ajudar famílias que enfrentam doenças raras.
O novo tratament, já aprovado no Japão desde 2021, permite que uma medicação administrada na veia atravesse a barreira sangue-cérebro e faça com que moléculas cheguem até o sistema nervoso central. O medicamento que disponibiliza a enzima deficiente nos pacientes com MPS II para todo o organismo, incluindo o sistema nervoso ainda está em análise pela Anvisa. A pesquisa clínica sobre o uso do alfapabinafuspe para o tratamento de pacientes com MPS II está sendo realizada no Brasil desde 2018.
Os resultados da Fase II revelaram que o tratamento pode ser benéfico para manter ou estabilizar o desenvolvimento neurocognitivo em pacientes com a forma grave da doença. Além disso, promove a melhora dos sintomas corporais e da atenção em pacientes com a forma atenuada, podendo ser usado para o tratamento de ambas as formas, tanto das manifestações neurológicas quanto dos sintomas que ocorrem em todos os outros órgãos dos pacientes, levando a falhas desses órgãos com o passar do tempo.
“Nos estudos clínicos, os indicadores de eficácia do medicamento foram bem evidentes, com redução dos biomarcadores da doença no sangue, na urina e no líquido que indica a atuação junto ao sistema nervoso central.” Giugliani apontou outros fatores positivos como melhora cognitiva, diminuição da medida do fígado e do baço e melhora da respiração que demonstraram a eficácia do tratamento, que fez grande diferença na qualidade de vida dos pacientes e dos parentes.